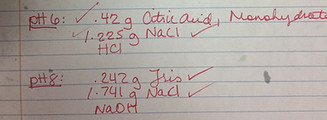Today, we collected our three optimization plates from the lab and found that our new method of keeping the water from splashing over was successful. We found that the sheet with the widest channels dried out the least, but all stayed relatively moisturized. All three sheets are pictured to the right. To see if our yeast grew, we decided to place the sheets under a black light which would theoretically cause the GFP in the yeast to fluoresce. Unfortunately, it appeared that the wildtype yeast we used did not have GFP in it. We knew, however, that the mutant yeast we made earlier in class has GFP in it. We tested the mutant yeast to see if it would fluoresce by pouring it onto our sheet and putting it under the light. It showed fluorescence (pictured to the right). We decided to redo the optimization using the same process as last time, only this time we used mutant yeast with GFP. This will allow us to discern growth using the black light, making it much easier to compare growth in the different channels. We also decided to optimize the amount of agar we should be plating in our channels to get optimal yeast growth. We designed a new wax print with eight channels of the same width, 0.25 inches. We wax printed it and used the same process we use to optimize the channel width, except this time changing the amount of agar in the channels. We checked how much agar it took to completely fill the channel and found that it took about 250 microliters. Knowing this, we decided to begin with 220 microliters of agar and go up by increments of 20 microliters, ending with 360 microliters of agar in the final channel (which, as can be seen in the picture to the right, spilled over the channel). Finally, we plated our 10 microliters of yeast in each channel and placed the sheets in Petri dishes to grow. We also received agar from group 2 with altered pHs of 2, 4, 6 and 8. They added various amounts chemicals and other ingredients, listed below, to create the acidic and basic solid agar (and are kind enough to share it with us). This will be used in our PDMS device.